Nanopolish 0.6
We’ve just pushed the latest version of nanopolish to github. This version has been long in development and has many changes:
- First official release of the methylation caller in the new
nanopolish call-methylationmodule. See here for a description of how to run it - Streamlined workflow for consensus and variant calling (
nanopolish eventalignno longer needs to be run) - You no longer need to provide the pore models files using the
--modelsargument. These are now compiled into the nanopolish binary, including the methylation models for R7.3, R9 and R9.4 - Initial support for FAST5 files generated by the Albacore basecaller
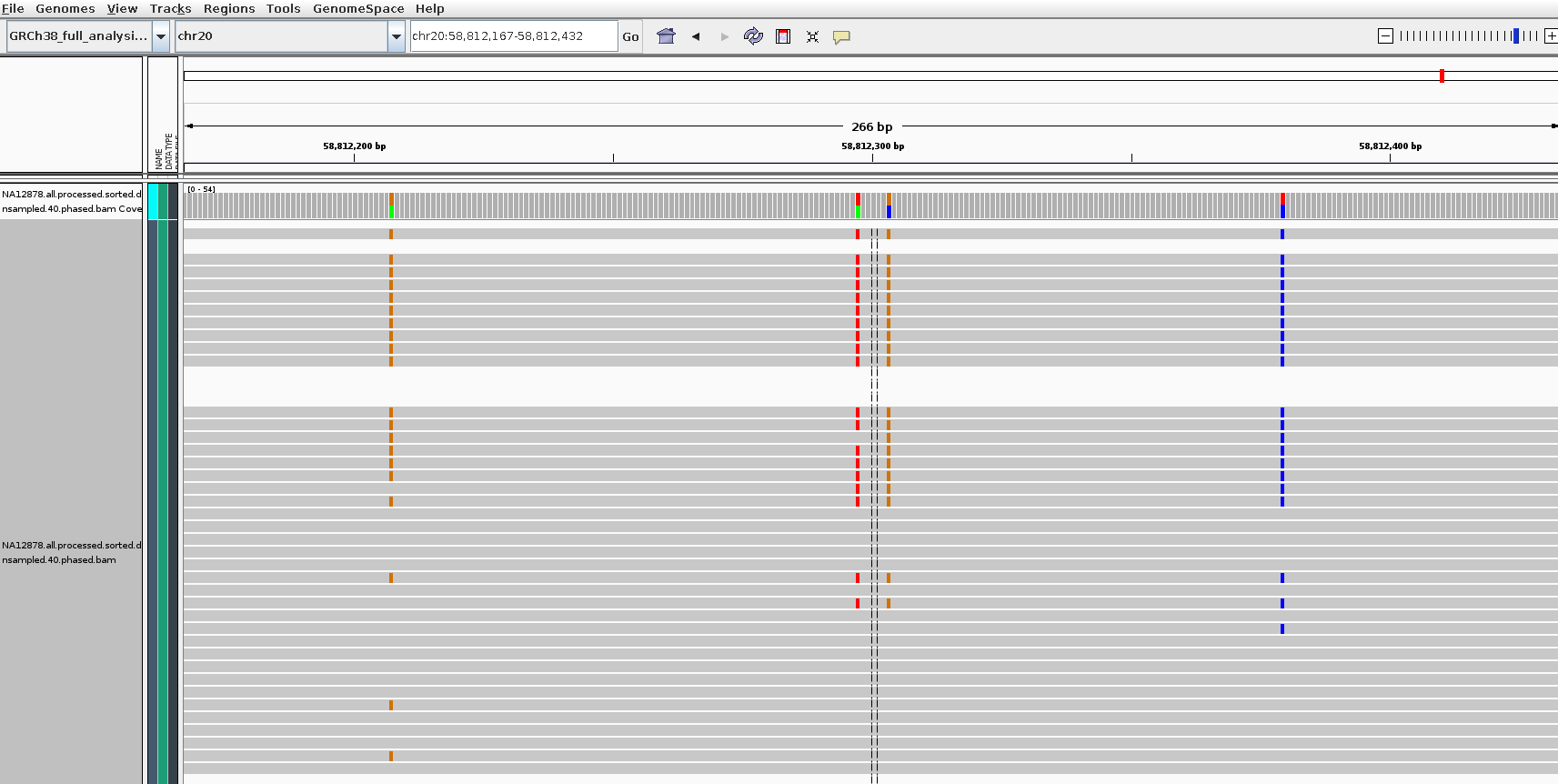
- A new experimental module,
nanopolish phase-reads, which calculates the set of variants each read supports from a provided VCF file. The output is a BAM file that can be used for phasing or clustering samples (more on this soon). Here’s a screenshot of the output:
Known Issues
The methylation caller is fairly lightweight as it only runs our expensive hidden Markov model at genomic CpG dinucleotides. The bottleneck in methylation detection currently is loading and parsing the signal-level information from the FAST5 files, not running the model. We’re working on optimizing this process.
If you have any comments or issues with this release please let us know in the comments here, on twitter or on github.